近日,清华大学物理系徐勇、段文晖研究组基于深度学习密度泛函理论哈密顿量(DeepH)方法,发展出一种具备第一性原理智能的深度学习电子结构计算方法DeepH-Zero。该方法首次在算法层面实现了神经网络与密度泛函理论的深度结合,赋予了模型基于物理原理的非监督学习能力。
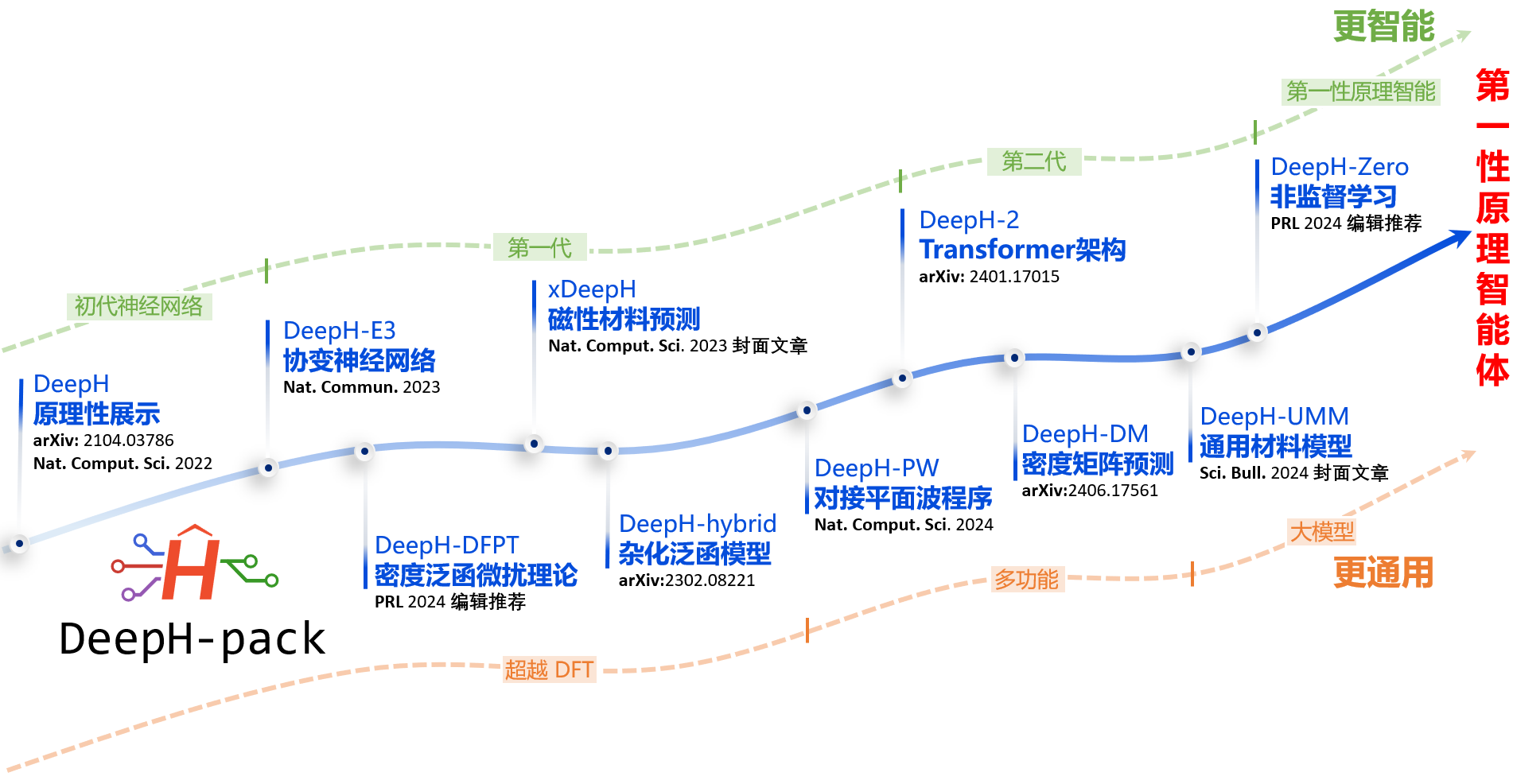
图1.DeepH 系列方法的发展历程
深度学习第一性原理计算是量子物理与人工智能的前沿交叉研究方向,可用于实现人工智能驱动的新物理与新材料发现。在前期研究中,徐勇、段文晖研究组发展了一种名为DeepH(Deep-learning DFT Hamiltonian)的深度学习电子结构计算方法。该方法能够从密度泛函(DFT)数据中学习,并准确预测给定材料的哈密顿量,进而高效计算多种材料性质。随后,团队分别基于等变网络技术,推出了DeepH-E3方法和xDeepH方法;通过引入Transformer架构,团队进一步提升了DeepH方法的通用性和泛化能力,推出了DeepH-2方法,并在此基础上发展出了DeepH通用材料模型。然而,先前研究普遍采用监督学习方法,深度学习与第一性原理算法彼此独立:即利用第一性原理计算构建数据集,再采用深度学习方法训练神经网络。未来发展需要将深度学习与第一性原理计算在理论与算法层面深度交叉融合,真正意义上实现具备第一性原理智能的深度学习科学计算。
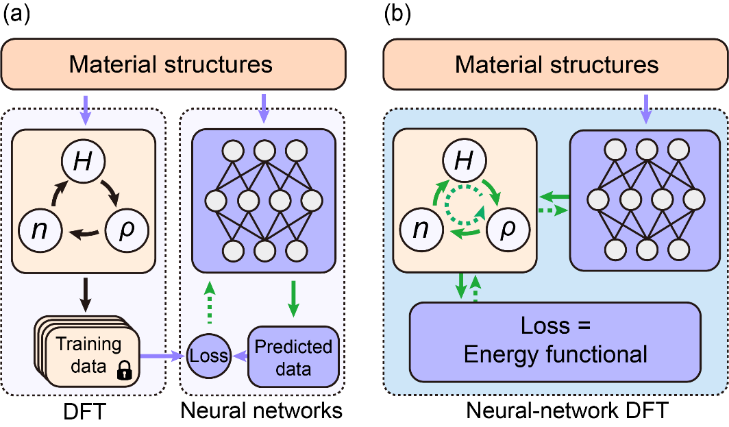
图2.监督学习(左图)与非监督学习(右图)。在前一类方法中,神经网络与密度泛函理论(DFT)的算法是分离的;后一类方法实现了两种算法的有机结合,被命名为“神经网络密度泛函理论”。DeepH-Zero首次展示了这类具备第一性原理智能的新理论方法
最新的研究进展中,着眼于深度学习与第一性原理计算的深度结合,团队研发了全新的DeepH-Zero方法,它能在零训练数据的情形下利用基本物理原理实现DeepH优化学习。该方法通过在神经网络中嵌入物理规则,巧妙地将神经网络与变分DFT算法结合,形成了一种名为“神经网络DFT”的无监督学习框架。DeepH-Zero在模型精度和泛化能力方面显著超越了传统监督学习框架,它能够在不依赖任何训练计算数据的情况下,实现对材料物性的精准预测。
在DeepH-Zero开发过程中,研究团队利用可微编程技术,从头构建了一整套拥有自主知识产权的新型DFT计算软件包——AI2DFT。通过自动微分和反向传播算法,AI2DFT程序能够高效执行DFT微分计算,实现了DFT程序与神经网络的无缝对接,进而实现基于物理原理的无监督学习。研究结果表明,通过将DFT基本原理融入神经网络设计,神经网络DFT在预测DFT物理量(如能量、电荷密度等)方面的精度超越了数据驱动方法。这项工作为深度学习与第一性原理计算的协同发展提供了崭新的思路,为实现具备第一性原理智能的深度学习电子结构计算发展开辟了新方向。
研究成果以“基于变分能量最小化的神经网络密度泛函理论”(Neural-network Density Functional Theory Based on Variational Energy Minimization)为题,于8月12日在线发表于《物理评论快报》(Physical Review Letters),并入选编辑推荐文章。这也是研究组今年在该方向发表的第二篇《物理评论快报》编辑推荐文章。
清华大学物理系教授徐勇和段文晖为该论文通讯作者,研究组“水木学者”博士后李洋、2023级博士生唐泽宸、2020级本科生陈泽洲为共同第一作者。合作者还包括研究组博士生孙明辉、赵柏恒、陶泓耕、袁子龙、李贺。研究得到基础科学研究中心、国家自然科学基金重点项目、国家科技部重点研发计划、北京市未来芯片技术高精尖创新中心、北京材料基因工程高精尖创新中心、清华大学“水木学者”、天津超算中心等项目单位的支持。
